その他の神経変性性認知症とその症状

アルツハイマー型認知症(ATD)に代表される脳の変性疾患では、特殊なタンパク質の蓄積や神経細胞の変性・脱落によって、脳が小さくなり、認知機能が低下します。この中でも、タウタンパクの異常蓄積を起こす疾患群を総称して「タウオパチー」といいます。ここでは、さまざまなタイプの「タウオパチー」を紹介します。記憶障害以外の症状が前面に出る疾患も多くあります。
- もくじ
- 非対称性のパーキンソニズム、失行、失語を伴う「大脳皮質基底核変性症(CBD)」
- パーキンソニズムと眼球運動障害が特徴「進行性核上性麻痺(PSP)」
- 舞踏運動などの不随意運動が特徴「ハンチントン病(HD)」
- 脊髄と脳の運動ニューロン疾患「筋萎縮性側索硬化症(ALS)」
- 軽度のアルツハイマー型と診断されやすい「嗜銀(しぎん)顆粒性認知症(AGD)」
- アルツハイマー型とピック病に似た症状を併発「石灰化を伴うびまん性神経原線維変化病(DNTC)」
非対称性のパーキンソニズム、失行、失語を伴う「大脳皮質基底核変性症(CBD)」
大脳皮質基底核変性症(CBD:corticobasal degenerarion)は、パーキンソン病の主症状(パーキンソニズム)と、大脳皮質症状を併せ持つ神経変性疾患です。前頭葉や頭頂葉を中心に左右非対称に萎縮し、脳回(大脳皮質にあるしわの隆起部分)が狭くなります。また、タウタンパクが神経細胞やグリア細胞に蓄積し、神経原線維変化が現れます。
パーキンソニズムは、筋肉が固くなる「筋強剛(きんきょうごう)」や「歩行障害」が多く、左右どちらかに強く現れます。
大脳皮質症状としては、基本的な動作に支障が出る「失行」、物体を認識できない「失認」などを発症します。認知症は必ずしも伴いませんが、認知症から発症することも少なくありません。人格変化や異常行動のほか、進行性の「失語」を伴うこともあります。
原因不明で、年齢、性別を問わず発症します。発病から2~3年で、パーキンソニズム、失語、失行などが現れます。
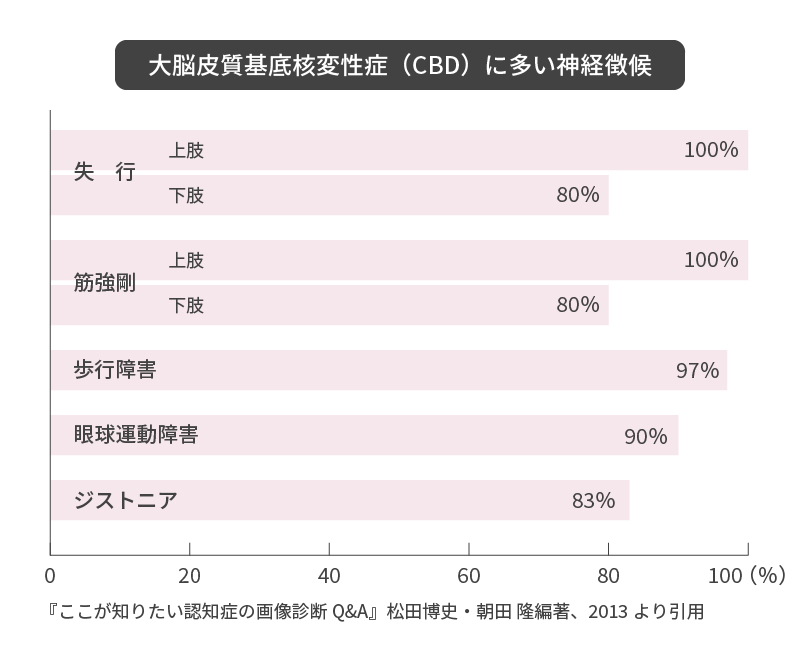 大脳皮質基底核変性症(CBD)の症状で最も目立つのは、運動機能に関係する神経徴候です。失行や筋強剛、歩行障害は、ほぼ全症例にみられ、持続性の筋収縮により、姿勢や動作が障害される「ジストニア」を発症する率も高くなっています。
大脳皮質基底核変性症(CBD)の症状で最も目立つのは、運動機能に関係する神経徴候です。失行や筋強剛、歩行障害は、ほぼ全症例にみられ、持続性の筋収縮により、姿勢や動作が障害される「ジストニア」を発症する率も高くなっています。
パーキンソニズムと眼球運動障害が特徴「進行性核上性麻痺(PSP)」
進行性核上性麻痺(PSP:progressive supranuclear palsy)は、中年期以降に発症する原因不明の神経疾患で、大脳皮質基底核変性症(CBD)の類縁といえる疾患です。パーキンソニズムと眼球運動障害の2つが、特徴的な症状です。
パーキンソニズムで特に目立つのが、バランスがとれずに、姿勢が不安定になったり、転びやすくなったりする「姿勢反射障害」です。また、動作が緩慢になり、体幹や首を中心に「筋強剛」が現れます。眼球が垂直方向に動かなくなる「眼球運動障害」では、特に下方向が見えづらくなります。言葉を正確に発音できない「構音障害」や「嚥下障害」も現れます。
通常は、進行とともに認知症が現れてきますが、初期には「アパシー(自発性の低下)」、「思考力低下」、「無気力」、「無関心」などがよくみられます。認知症症状が先に発症した場合、人格変化や行動異常が中心に現れる症例も報告されています。
脳の萎縮は、大脳基底核のほか、大脳の下にある脳幹、小脳などにも現れます。また、神経細胞やグリア細胞に、タウタンパクの過剰な蓄積が見られます。
発症は40歳以降、特に60歳代の男性に多く、原因不明で、孤発性が中心です。まれに遺伝性もあります。運動機能が目立って低下していきますが、その他の症状は徐々に進行します。発症から5~10年で死に至ります。
進行性核上性麻痺(PSP)と大脳皮質基底核変性症(CBD)は類縁疾患
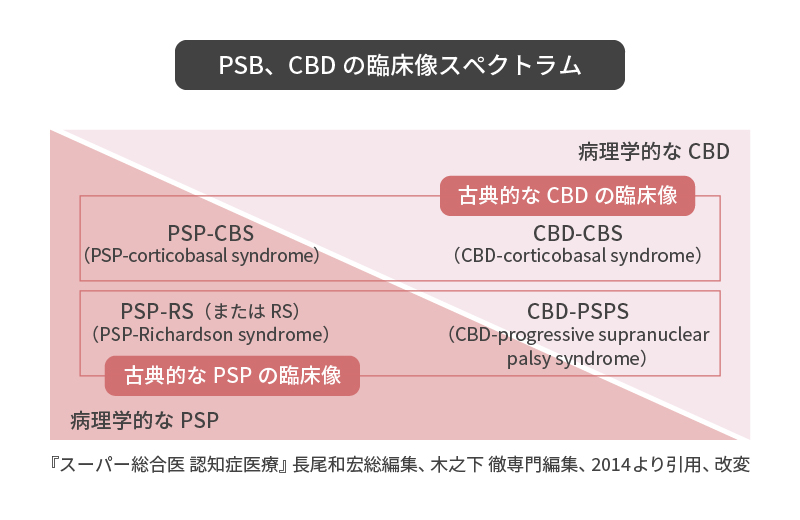 進行性核上性麻痺(PSP)と大脳皮質基底核変性症(CBD)は連続した疾患です。病理学的にどちらかに診断されても、その臨床像は単一ではありません。それぞれに典型例とそうでない例があり、上図のとおり、主に4タイプに分類されます。
進行性核上性麻痺(PSP)と大脳皮質基底核変性症(CBD)は連続した疾患です。病理学的にどちらかに診断されても、その臨床像は単一ではありません。それぞれに典型例とそうでない例があり、上図のとおり、主に4タイプに分類されます。
舞踏運動などの不随意運動が特徴「ハンチントン病(HD)」
ハンチントン病(HD:Hungtinton’s disease)は、大脳の深部にある大脳基底核、特に線条体と淡蒼球が萎縮し、「舞踏運動」、「精神症状」、「認知症」を生じる遺伝性の変性疾患です。
多くは中年期で発症しますが、20歳前に発症する若年型もあります。また、世代を経るうちに発症年齢が早まる「表現促進現象」が確認されています。
舞踏運動とは、自分の意思とは無関係に生じる不規則ですばやい運動で、手先や舌、口などに現れる。精神症は、「不安」、「うつ気分」、「易刺激性(興奮しやすい)」、「アパシー(自発性の低下)」などがみられます。また、自殺のリスクも比較的高いといわれています。
通常は、舞踏運動で発症しますが、それ以前に、軽度ながら、精神症状や認知機能低下が認められるという報告もあります。
脊髄と脳の運動ニューロン疾患「筋萎縮性側索硬化症(ALS)」
筋萎縮性側索硬化症(ALS:amyotrophic lateral sclerosis)は、脳からの運動指令を伝える運動ニューロンが、選択的に障害される変性疾患です。
40〜60歳代の男性に多い疾患で、90%以上は孤発性です。
手の脱力からはじまり、進行すると、手足がまったく動かせなくなります。全身の「筋力低下・筋肉萎縮」により、「言語障害・構音障害」なども現れます。末期になると呼吸筋も強く萎縮するため、人工呼吸器の使用が必要になります。
まれに認知症を発症しますが、特に目立つのが、「行動異常」、「自発性や意欲の低下」といった、前頭葉機能の低下です。
前頭側頭葉変性症(FTLD)と同じく、TDP-43というタンパク質の異常蓄積がみられることから、同疾患との関連性も指摘されています。
軽度のアルツハイマー型と診断されやすい「嗜銀(しぎん)顆粒性認知症(AGD)」
嗜銀顆粒性認知症(AGD:argyrophilic grain disease)は、銀を用いた組織染色で反応する「嗜銀顆粒」が現れる変性疾患で、一種の加齢性変化とされています。リン酸化したタウタンパクが神経細胞の樹状突起などに蓄積し、萎縮が側頭葉内側から海馬を経て、前頭葉にまで広がります。
発症頻度はアルツハイマー型認知症(ATD)に次ぐともいわれますが、認知度が低く、画像や症状からは確定できないため、軽度アルツハイマー型と診断されやすい傾向があります。アルツハイマー型との合併例も多く、アルツハイマー型の病状を促進する可能性も指摘されています。
進行が遅く、軽度認知障害(MCI)程度の記憶障害が続きます。「易刺激性(興奮しやすい)」、「アパシー(自発性の低下)」などの前頭葉症状が現れることもあります。
アルツハイマー型とピック病に似た症状を併発「石灰化を伴うびまん性神経原線維変化病(DNTC)」
石灰化を伴うびまん性神経原線維変化病(DNTC:diffuse neurofibrillary tangles with calcification)は、比較的まれな神経疾患で、神経原線維変化と大脳基底核の石灰化が特徴です。
タウタンパクの蓄積により、神経原線維変化が多数出現しますが、老人斑はみられません。また、レビー小体型と同様、αシヌクレインの異常沈着も認められます。
初期には、アルツハイマー型認知症(ATD)のような「記憶障害」、「見当識障害」の症状が多く、進行とともに「易刺激性」、「攻撃性」、「無関心」など、前頭側頭型認知症(FTD:ピック病と同義)に似た症状が加わります。
なお、石灰化を伴わず、神経原線維変化が単独で現れる病態もあります。これは「神経原線維変化老年期認知症(SD-NFT)」、または「神経原線維変化優位型認知症」と呼ばれています。


